Algorithms for Characterizing Copy Number Variation in Human Genome
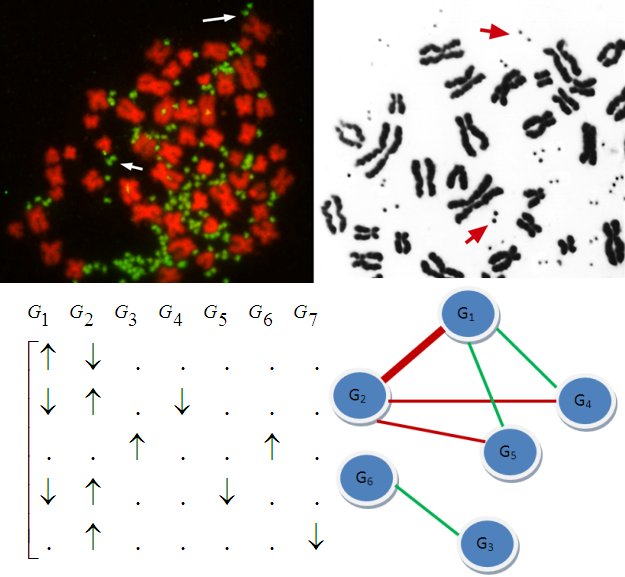
Not long ago, it was discovered that individuals may differ in copy
numbers of their genes,
meaning that a segment of DNA may have more or less copies than usual
in an individual's chromosome. Recent research suggests that
these variations are associated with many diseases including Autism
and Schizophrenia. Copy number
variation (CNV) in somatic cells also underly various cancers. Copy
numbers are usually identified using SNP microarrays, however,
short-read sequence data is emerging as an important resource
for characterizing structural variation in human genome.
As an interdisciplinary group of researchers at
Case
Western Reserve University, we develop optimization based algorithms for fast and accurate identification
of rare and de novo CNVs, as well as copy number polymorphisms (CNPs) and
other forms of structural variation from these two data sources, with a view
to enabling personalized genomics applications.
Software
- COKGEN:
An R package for optimization-based identification of rare and de novo
copy number variants from SNP microarray data.
Please stay tuned for POLYGON, an R package for identification of copy
number polymorphisms from SNP microarray data. POLYGON is designed to work with
any CNV identification algorithm to consolidate and improve their findings
by drawing strength from multiple samples in the population.
Publications
- G. Yavas, M. Koyuturk, and T. LaFramboise.
Optimization algorithms for identification and genotyping of copy number polymorphisms
in human populations. 5th IAPR Int'l Conf. on Pattern
Recognition in Bioinformatics (PRIB'10), in press.
- G. Yavas, M. Koyuturk, M. Ozsoyoglu, M. P. Gould, and T. LaFramboise.
COKGEN:
A software for the identification of rare copy number variation from SNP microarrays.
Pacific
Symposium on Biocomputing (PSB'10), 371-382, 2010.
- G. Yavas, M. Koyuturk, M. Ozsoyoglu, M. P. Gould, and T. LaFramboise.
An optimization
framework for unsupervised identification of rare copy number variation from SNP array
data.
Genome Biology, 10:R119, 2009.
People

Katie Wilkins
Undergraduate Student, Computer Science/Biochemistry

Matthew M. Ruffalo
Ph.D. Student, Electrical Engineering & Computer Science

Gokhan Yavas
Ph.D. Student, Electrical Engineering & Computer Science

Assistant Professor,
Genetics

Assistant Professor,
Electrical Engineering & Computer Science
Acknowledgments
This project is supported
by National Science Foundation
Award IIS-0916102.
| Not long ago, it was discovered that individuals may differ in copy numbers of their genes, meaning that a segment of DNA may have more or less copies than usual in an individual's chromosome. Recent research suggests that these variations are associated with many diseases including Autism and Schizophrenia. Copy number variation (CNV) in somatic cells also underly various cancers. Copy numbers are usually identified using SNP microarrays, however, short-read sequence data is emerging as an important resource for characterizing structural variation in human genome. As an interdisciplinary group of researchers at Case Western Reserve University, we develop optimization based algorithms for fast and accurate identification of rare and de novo CNVs, as well as copy number polymorphisms (CNPs) and other forms of structural variation from these two data sources, with a view to enabling personalized genomics applications. |
|
Software
- COKGEN: An R package for optimization-based identification of rare and de novo copy number variants from SNP microarray data.
Publications
- G. Yavas, M. Koyuturk, and T. LaFramboise. Optimization algorithms for identification and genotyping of copy number polymorphisms in human populations. 5th IAPR Int'l Conf. on Pattern Recognition in Bioinformatics (PRIB'10), in press.
- G. Yavas, M. Koyuturk, M. Ozsoyoglu, M. P. Gould, and T. LaFramboise. COKGEN: A software for the identification of rare copy number variation from SNP microarrays. Pacific Symposium on Biocomputing (PSB'10), 371-382, 2010.
- G. Yavas, M. Koyuturk, M. Ozsoyoglu, M. P. Gould, and T. LaFramboise. An optimization framework for unsupervised identification of rare copy number variation from SNP array data. Genome Biology, 10:R119, 2009.
People
|
Katie Wilkins Undergraduate Student, Computer Science/Biochemistry |
|
Matthew M. Ruffalo Ph.D. Student, Electrical Engineering & Computer Science |
|
Gokhan Yavas Ph.D. Student, Electrical Engineering & Computer Science |
|
Assistant Professor, Genetics |
|
Assistant Professor, Electrical Engineering & Computer Science |